https://www.researchgate.net/publication/267876438_Genomic_Structure_in_Europeans_dating_back_at_least_36200_years
8 NOVEMBER 2014 •VOL 346 ISSUE 6213 1113
Genomic structure in Europeans dating back at least 36,200 years
Andaine Seguin-Orlando, 1
*Thorfinn S. Korneliussen, 1
*Martin Sikora, 1
*Anna-Sapfo Malaspinas, 1
Andrea Manica, 2
Ida Moltke, 3,4
Anders Albrechtsen, 4
Amy Ko, 5
Ashot Margaryan, 1
Vyacheslav Moiseyev, 6
Ted Goebel, 7
Michael Westaway, 8
David Lambert, 8
Valeri Khartanovich, 6
Jeffrey D. Wall, 9
Philip R. Nigst, 10,11
Robert A. Foley, 1,12
Marta Mirazon Lahr, 1,12 †
Rasmus Nielsen, 5 †
Ludovic Orlando, 1
Eske Willerslev, 1 †
1 Centre for GeoGenetics, Natural History Museum of Denmark, University of Copenhagen, Øster Voldgade 5–7, 1350 Copenhagen, Denmark.
2 Department of Zoology, University of Cambridge, Downing Street, Cambridge, CB2 3EJ, UK.
3 Department of Human Genetics, University of Chicago, 920 East 58th Street, Cummings Life Science Center, Chicago, IL 60637, USA.
4 The Bioinformatics Center, University of Copenhagen, Ole Maaløes Vej 5, 2200 København N, Denmark.
5 Department of Integrative Biology, University of California, Berkeley, CA 94720, USA.
6 Department of Physical Anthropology, Kunstkamera, Peter the Great Museum of Anthropology and Ethnography, Russian Academy of Sciences, 24 Srednii Prospect, Vassilievskii Island, St. Petersburg, Russia.
7 Center for the Study of the First Americans and Department of Anthropology, Texas A&M University, TAMU-4352, College Station, Texas 77845-4352, USA.
8 Environmental Futures Research Institute, Griffith University, 170 Kessels Road, Nathan, Brisbane, Queensland 4111, Australia.
9 Department of Epidemiology and Biostatistics, University of California San Francisco, 185 Berry Street, Lobby 5, Suite 5700, San Francisco, CA 94107, USA.
10 Division of Archaeology, University of Cambridge, Cambridge, Downing Street, CB2 3DZ, UK.
11 Department of Human Evolution, Max-Planck-Institute for Evolutionary Anthropology, Leipzig, Deutscher Platz 6, D-04103, Germany.
12 Leverhulme Centre for Human Evolutionary Studies, Department of Archaeology and Anthropology, University of Cambridge, Cambridge, Fitzwilliam Street, CB2 1QH, UK.
*These authors contributed equally to the work.
†Corresponding author.
The origin of contemporary Europeans remains contentious.We obtained a genome sequence from Kostenki 14 in European Russia dating from 38,700 to 36,200 years ago, one of the oldest fossils of anatomically modern humans from Europe. We find that Kostenki 14 shares a close ancestry with the 24,000-year-old Mal’ta boy from central Siberia, European Mesolithic hunter-gatherers, some contemporary western Siberians, and many Europeans,but not eastern Asians. Additionally,the Kostenki 14 genome shows evidence of shared ancestry with a population basal to all Eurasians that also relates to later European Neolithic farmers. We find that Kostenki 14 con-tains more Neandertal DNA that is contained in longer tracts than present Euro-peans. Our findings reveal the timing of divergence of western Eurasians and East Asians to be more than 36,200 years ago and that European genomic structure today dates back to the Upper Paleolithic and derives from a metapopulation that at times stretched from Europe to central Asia.
The ancestors of contemporary Eurasians are believed to have left Africa some 60,000 to 50,000 years ago (60 to 50 ka) (1,2), possibly 30,000 to 40,000 years later than Australo-Melanesian ancestors (3). Despite controversies about routes out of Africa, the first Upper Paleolithic (UP) industries of Eurasia are found in the Levant from ~48 ka (4,5). Expansion into Europe took place through multiple events that by ~40 ka had generated a spatially and culturally structured anatomically modern human (AMH) population — from Russia (6) to Georgia (7), Bulgaria (8), southern Europe (9,10), and the United Kingdom (11). The few AMH fossils associated with these initial UP industries are morphologically variable (9,12–17). In western Eura-sia, the distinctive Aurignacian toolkit, first observed at Willendorf (Austria) by 43.5 ka (18), became predominant across the earlier range by 39 ka. Although analyses of ancient human genomes have advanced our understanding of the European past, revealing contributions from Paleolithic Siberians, European Mesolithic, and Near Eastern Neolithic groups to the European gene pool (19–23), the possible contri-bution of the earliest Eurasians to these later cultures and to contemporary human populations remains unknown. To investigate this, we sequenced the genome of Kostenki 14 (K14, Markina Gora) (Fig. 1A).
The locality of Kostenki-Borshchevo on the Middle Don River, Russia, has one of the most extensive Paleolithic records in eastern Europe.
The K14 human skeleton was excavated in 1954 (24) and was recently dated to 33,250 T500 radiocarbon years before the present (B.P.) (25),38.7 to 36.2 thousand calendar years B.P. (ky cal B.P.), in agreement with the stratigraphic position of the burial that cuts into the Campanian Ignimbrite ash layer dated to ~39.3 ky cal B.P. (26). Below the skeleton, there is a distinctive early UP industry, with end scrapers, burins, prismatic cores,and bone artifacts (layer IV);the cultural layer above (layer III) has a regionally local character (27,28) [supplementary materials (SM) S1 and S2].
We performed 13 DNA extractions from a total of 1.285 g of the left tibia (dorsal side of the shaft), using two extraction methods based on silica purification (29,30). We first constructed seven Illumina libraries and validated the presence of typical signa-tures of postmortem DNA damage, using a fraction of DNA extracts (SM S3). The remaining extracts were built into 63 libraries after enzymatic uracil-specific excision reagent (USER) treatment to limit the effect of nucleotide misincorporations in down-stream analyses (31) (table S2). Additionally, a limited fraction of two DNA extracts was purified for methylated DNA fragments using methyl binding domain (MBD) enrichment (32) before USER treatment and library building,for a total of eight DNA libraries. Following stringent quality criteria for read alignment, we identified a total number of 175.2 million unique reads aligning against the human reference genome hg19, representing an average depth of coverage of 2.84X (SM S4). The eight USER-treated DNA libraries that exhibited limited error rates and contamination levels were selected for further analyses. This restricted the data set to 148.9 million unique reads, representing a final depth of coverage of 2.42X. We exploited the fact that K14 was a male and used the heterozygosity levels present in the X chromo-some to estimate overall levels of contamination around 2.0% (SM S5 and S6 and table S5).
The population genetics analyses results are robust to contamination of that level. In particular, we replicated the main analyses with selected libraries with varying conta-mination levels and observed no qualitative effect on the results (see SM S9 for details).
Mitochondrial analyses confirmed the sequence previously reported for K14 [haplo-group U2 (33)],which supports data authenticity. The Y chromosome belongs to hap- logroup C M130, the same as in La Braña - a late Mesolithic hunter-gatherer (MHG) from northern Spain (22) (SM S7).
To identify patterns of shared ancestry and admixture among K14, other ancient genomes and contemporary Eurasians [based on a single-nucleotide polymorphism (SNP) array panel of 2091 individuals from 167 populations], we carried out a series of analyses - model-based clusterring and principal component analysis (PCA) - to show the contribution of diverse genetic components within K14: D statistics to ex-plore the affinity of K14 to pairs of populations (using Mbuti Pygmy as an outgroup); f4 statistics to test whether a given modern population is equidistant to an ancient individual and a particular recent group (here, Sardinians), given an outgroup (here, Papuans); and f3 statistics to explore both patterns of admixture (“admixture” f3) and shared ancestry (“outgroup” f3). Key results were also replicated using two whole-genome sequencing data sets of modern individuals from worldwide populations (23,34).
Model-based clustering analyses (35) show that K14 has different genetic compo-nents of substantial size (Fig. 1B and SM S10), suggesting the sharing of sets of alleles with different Eurasian groups. The largest fraction of K14’s ancestry derives from a component that is maximized in European MHGs and also predominant in contemporary northern and eastern Europeans. The genetic affinity of K14 to con-temporary Europeans is also observed using outgroup f3 statistics (36). Using Mbuti Pygmy as an outgroup, we find that among a panel of 167 contemporary popu- lations, Europeans have the greatest affinity (i.e., the largest f3) to K14 (Fig. 1C).
This conclusion is also formally supported by comparing pairs of populations to K14 using the D statistics of the form D (Mbuti Pygmy, K14; Population 1, Population 2). This statistic is expected to be equal to zero if K14 is symmetrically related to Popu-lation 1 and Population 2,whereas its expectation is negative (positive) if K14 is more closely related to Population 1 (Population 2). For pairs of populations involving East Asians (Population 1) and Europeans (Population 2), K14 is always significantly more closely related to Europeans [e.g., Z = 12.1 (Han and Lithuanians)], in all data sets analyzed (SM S9 and table S7).We also confirmed that these results are robust to possible contamination from a modern DNA source by filtering for reads with a high likelihood of ancient DNA using a model-based approach (37), as well as calculating contamination-corrected Dstatistics (23) (SM S9 and fig. S18).
Within Europe, northern Europeans show the closest affinity to K14, based both on the f3 (Fig. 1D) and Dstatistics [e.g., Z = 6.7 for Sardinians and Lithuanians; table S7 and fig. S16]. This pattern closely resembles that of European MHGs (La Braña, Ajv58, Loschbour, and Motala) and Mal’ta (MA1) (figs.S14 and S15), with the excep- tion of the latter’s strong genetic affinity with Native Americans, which is unique to that individual.
Furthermore, a direct comparison to ancient genomes in the outgroup f3 statistics shows that K14 has a higher affinity with MHGs (Loschbour and La Braña) than any other ancient individual or contemporary population (fig. S14). Together with the rare Y chromosome lineage shared with La Braña, these results provide strong evidence
of shared ancestry and extensive gene flow between UP West Eurasian people related to K14 and European MHGs and their contemporary European descendants.
An interpretation of the above results would be that K14 is an early member of a li-neage leading to western Eurasian MHGs after their split from the proposed ancest-ral northern Eurasian lineage, including MA1. However, D statistics of the form D (Mbuti Pygmy, Modern; Ancient,K14) - which test whether K14 and an ancient indivi- dual form a clade with respect to a modern population - reject this simple tree-like re-lationship. We find that all contemporary non-Africans, except Australo-Melanesians, are closer to either MA1 or MHGs than to K14 [e.g., Z = –5.3 for D (Mbuti and Han; Loschbour and K14); SM S9, table S10,and fig.S19]. This would suggest a basal po-sition of K14 with respect to MHGs and ancient north Eurasians, which is also shown in admixture graphs using TreeMix (SOM S12 and figs. S24 and S25). In addition, a sizeable component of K14’s ancestry observed in the model-based clustering ana-lyses is predominant in contemporary Middle Eastern/Caucasus (ME/C) populations and Neolithic ancient genomes (NEOL) (Gok2, Iceman, and Stuttgart) but absent in MA1 or MHGs (Fig. 1B and fig. S20).
This component has been associated with a suggested “basal Eurasian” lineage contributing to NEOL to explain an observed increase in allele sharing between MHGs / MA1 and East Asians compared with NEOL (21). Because K14 shows the same pattern as NEOL, a parsimonious explanation would be that K14 also derives some ancestry from a related basal Eurasian lineage.
Consistent with this hypothesis, we find that East Asians are equally distant to NEOL and K14, using D statistics as described above [e.g., Z= 0.0 for D (Mbuti, Han; Stuttgart, K14); tables S10 and S11].
This suggests that the main ancestral components proposed for contemporary Euro-pea ns, including the Middle Eastern component commonly attributed to the expan-sion of early farmers within Europe, were likely already genetically differentiated and related through complex gene flow by the time of K14, at least 36.2 ka (Fig. 2).
We further investigated the relationship of K14 and the other ancient genomes to East Asian and Siberian populations using f4 statistics f4 (Sardinian, Ancient; Modern, Papuan), which measure whether a modern population shares more alleles with contemporary Europeans or an ancient genome.
We find that all Siberian and East Asians are equally distant from western MHGs (all |Z|<1.9) (Fig. 3D and table S12), supporting the postulated early split between East Asians and western Eurasians. In contrast to MHGs and MA1, all Siberian popula-tions are genetically closer to contemporary Europeans (Sardinians) than to K14 (3.1 < |Z| < 9.9) (table S12), particularly those from the Yenisei and Ob’ basins (e.g., Shors,Z = 8.0) (Fig.3A). Furthermore,these populations derive parts of their ancestry from a European hunter-gatherer (HG) component inferred in the clustering analysis (Fig. 1D and fig. S20),with populations showing a higher HG ancestry proportion also being closer to contemporary Europeans,using the f4 statistic (Spearman r = 0.96; P = 3.0 × 10 −18) (Fig. 3D and table S13).
Notably, the opposite pattern is observed with Scandinavian MHGs (Ajv58 and Mota-la), where the same populations tend to share more alleles with MHGs than contem-porary Europeans and the HG component is negatively correlated with f4 (e.g., Mo-tala r=–0.85; P=6.2×10 −10) (Fig. 3, C and D). Calculating admixture f3 statistics, we find significant evidence for admixture in those populations, with a variety of Siberian and European source populations. The best pair of source populations (i.e.,the most negative f3 statistic) involves Swedish MHGs (Motala) and Evens (a northeast Sibe-rian population) [e.g., f3 (Shors; Evens, Motala) = –0.012; Z = –9.1] (table S14).
Altogether,these results suggest that contemporary Siberian populations from the Yenisei basin derive part of their gene pool from a Eurasian HG population that shares ancestry with K14 but is more closely related to Scandinavian MHGs than to either MA1 or western European MHGs,indicating gene flow between their ancestors and Scandinavian Europe after K14 but before the Mesolithic (between 36.3 and 7 ky B.P.).
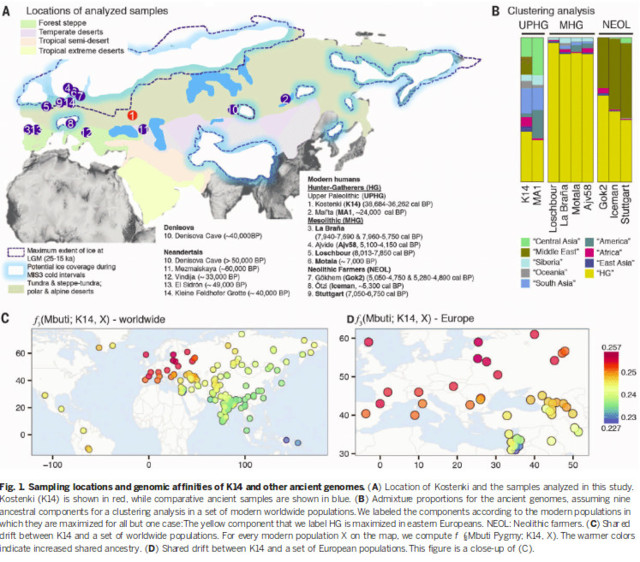
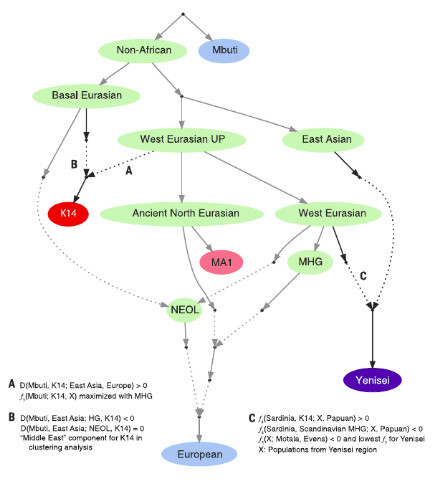
Fig. 2. Relationships of the K14 sample and MA1, MHG, NEOL, modern Europeans, and the modern populations in the Yenisei region. his representation is a possible to-pology consistent with the results presented in this study in the context of the rela-tionships described by Lazaridis et al.(21) for the modern European populations and Raghavan et al.(23) for MA1. Present-day populations are colored in blue, ancient poplation in red, and ancestral populations in green. Solid lines represent descent without admixture events, and dashed lines show admixture events. Arrows do not depart from ancient samples (K14 and MA1) because they represent relationships of population ancestry. We only show the topology of the potential population tree: There is no notion of time in this representation.The tree is not the result of a model-fitting procedure but rather a possible topology consistent with the key results (A, B, and C) of this study.
Kommentit